TP 4: Simulation und Theorie der Strukturbildung (1. FP)
Projektleiter: Prof. Dr. Karl Heinz HoffmannTU Chemnitz, Professur für Computerphysik, Chemnitz
Projektziele:
- Verständnis der Strukturbildung - Schließen einer Lücke

- Verständnis für die grundlegenden Prozesse der Morphologie- und Texturbildung von Nanokompositen
- Bestimmung von vergröberten Wechselwirkungsmodellen
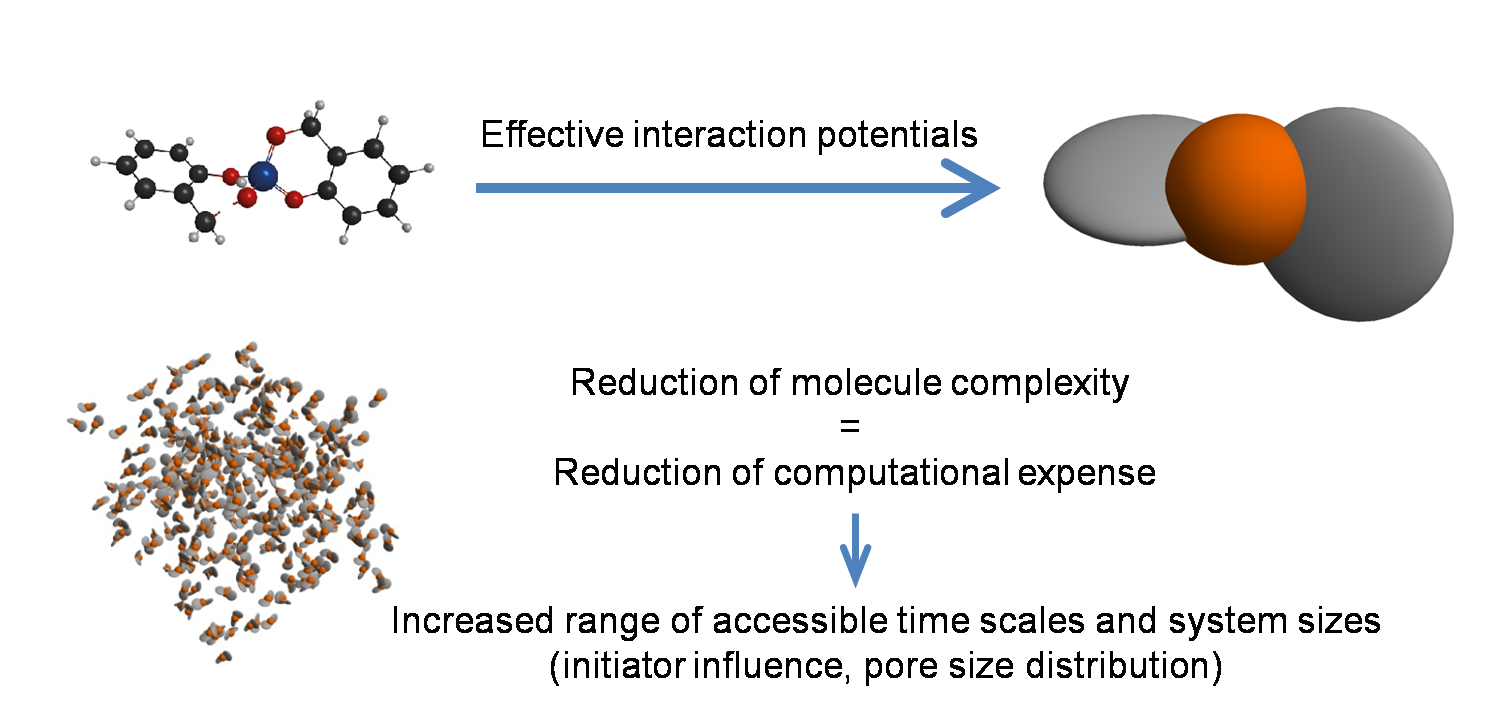
- Feststellung Einfluss des Monomeraufbaus und der resultierenden räumlichen Struktur auf die entstehenden Nanostrukturen
Atomare Molekulardynamik mit reaktiven Kraftfeldern
- Adaption sowie Parametrisierung und Weiterentwicklung des reaktiven Wechselwirkungskraftfeldes ReaxFF für Molekulardynamiksimulationen. ReaxFF ist ein Bond-Order-Potential, bei dem die Ordnung der Atombindung über den Atomabstandes zwischen jeweils einem Paar von Atomen bestimmt wird. Das Brechen bzw. Bilden von Atombindungen wird über eine Bindungslänge zu Bindungsordnung Relation dargestellt.
- Erstmalige Adaption vorhandener ReaxFF-Kraftfeld-Parametrisierung für das Zwillingsmonomer 2,2'-spirobi[4H-1,3,2-benzodioxasiline](1). (Farbschema: Si - gelb, O - rot, C - grau, H - weiß)
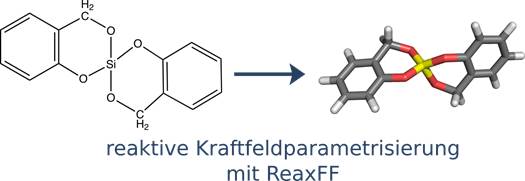
- Modellierung des im Rahmen von quantenchemischen Berechnungen (TP1 und J.Friedrich) vorhergesagten Reaktionspfades von (1) mit bereits vorhanden ReaxFF-Parametrisierungen ist auf Grund der Komplexität des Zwillingspolymerisationsprozesses nicht realisierbar.
- Entwicklung von eigenen ReaxFF-Parametrisierungen zur Modellierung der Zwillingspolymerisation.
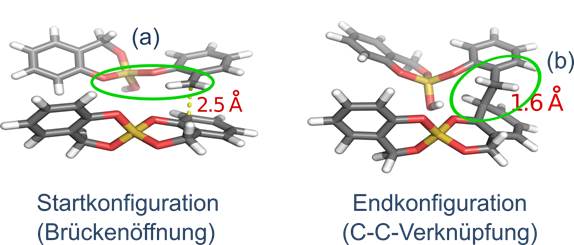
- Entwicklung einer ersten ReaxFF-Parametrisierung zur Modellierung der Bildung des organischen Netzwerkes unter der Annahme, dass nur das Proton der Säure der Initiator zu berücksichtigen ist. Mit dieser TP-ReaxFF-Parametrisierung gelang erstmalig eine erfolgreiche Simulation der aufeinanderfolgenden Reaktionsschritte (a) die Brückenöffnung und (b) die C-C-Verknüpfung der organischen Komponente, wie quantenchemisch berechnet und experimentell angenommen. (Farbschema: Si - gelb, O - rot, C - grau, H - weiß)
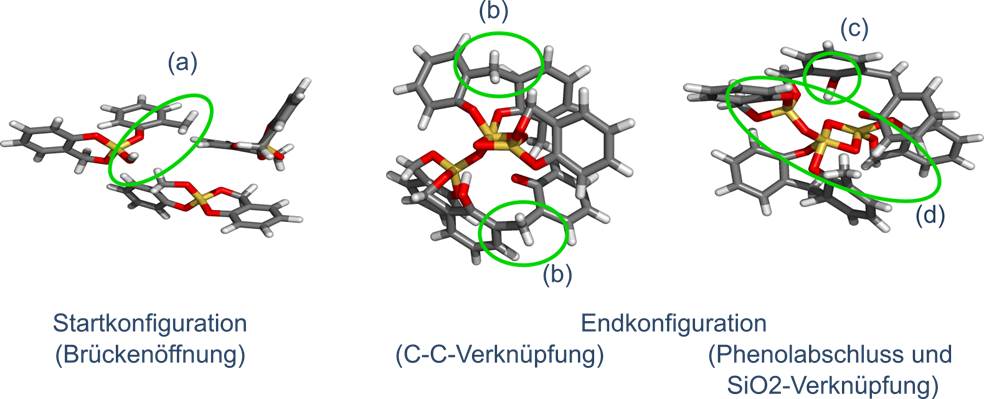
- Auf Grund der Komplexität der Reaktion der Zwillingspolymerisation ist es notwendig das Konzept von ReaxFF zu erweitern und verschiedene Atomtypen für charakteristische chemische Eigenschaften des Zwillingsmonomers einzuführen. Im Rahmen der sdar-ReaxFF-Parametrisierung ist es gelungen für ein kleines System (a) die Brückenöffnung, (b) die C-C-Verknüpfung, (c) die SiO2-Verknüpfung und (d) den Phenolabschluss direkt aufeinanderfolgend zu simulieren. Dies entsprach den quantenchemischen Berechnungen. Jedoch wurde weiterhin nur der Protonen-Einfluss auf den Reaktionsmechanismus berücksichtigt. (Farbschema: Si - gelb, O - rot, C - grau, H - weiß)
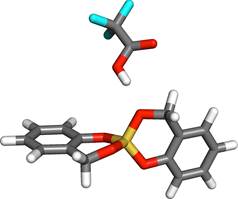
- Es zeigte sich, dass bei der Simulation des Zwillingspolymerisationsprozesses für größere System die vollständige Säure parametrisiert werden muss, so dass alle Wechselwirkungen, u.a. auch die Coloumb-Wechselwirkung korrekt berücksichtig werden. Hierbei konzentrierten wir uns zunächst auf die Triflouressigsäure (TFA). Die für die Anpassung der sdar-ReaxFF-Parametrisierung für TFA notwendigen quantenchemischen Daten wurden von der Gruppe um J.Friedrich gerechnet und werden aktuell in die Parametrisierung integriert. Eine mögliche Startkonfiguration sehen sie im Bild (Farbschema: F - blau, Si - gelb, O - rot, C - grau, H - weiß).
Vergröberte Molekulardynamik mit vergröberten reaktiven Kraftfeldern
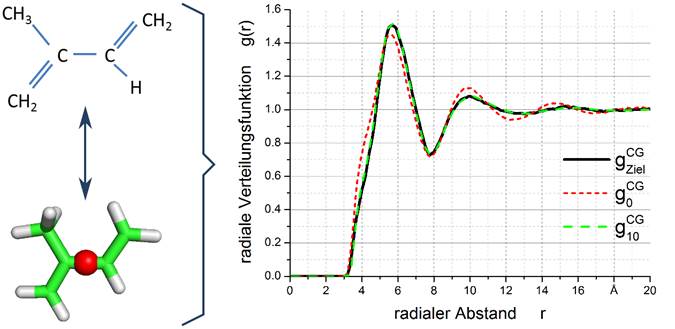
- Es wurden erste Simulationstestreihen durchgeführt, ob das Simulationswerkzeug IBIsCO für die Zwillingspolymerisation verwendet werden kann. Dies wurde zunächst an den einfachen Strukturen Isopren (siehe Bild) und Polyisopren getestet. Es zeigte sich, dass IBIsCO eine hohe Qualität der Strukturwiedergabe der vergröberten Strukturen im Vergleich zur atomaren Struktur hat und sich daher für die Zwillingspolymerisation sehr gut eignet. Im Bild ist die Strukturformel von Isopren, das Mapping-Schema von IBIsCO von der räumlichen Struktur (grün-weiße Struktur) auf die vergröberte Perlen-Struktur (rote Perle im Zentrum der Struktur) und die radiale Verteilungsfunktion von Isopren (schwarz) im Vergleich zu der der vergröberten Struktur (grün-gestrichelte Linie).